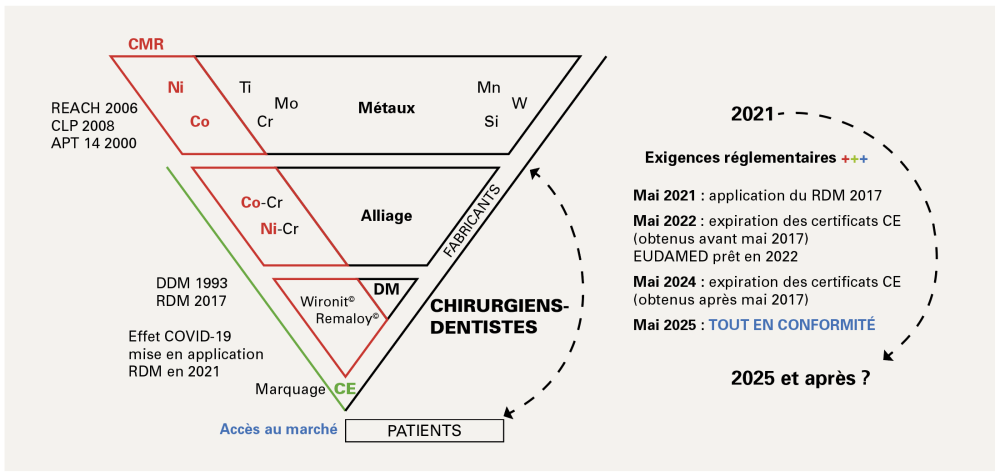

Le 22 septembre 2017, à la surprise d’un grand nombre de professionnels de santé et d’industriels du domaine de la dentisterie, le comité d’évaluation des risques (CER) de l’Agence européenne des produits chimiques (ECHA) a adopté un avis pour réviser la classification et l’étiquetage du cobalt (Co) métallique [1]. Depuis 2020, le Co métallique est ainsi devenu une substance classée cancérigène, mutagène et toxique pour la reproduction (CMR). Ceci n’est pas sans conséquence dans notre pratique quotidienne, notamment en raison de la mise en place progressive (2017-2025) du nouveau règlement (UE) 2017/745 sur les dispositifs médicaux (RDM) [2]. Ce RDM impose de nouvelles exigences envers les dispositifs médicaux contenant une substance classée CMR. Le dispositif médical ne peut alors être mis sur le marché que si aucune alternative n’est disponible et que si une justification est fournie par le fabricant ou par l’organisme notifié (ON) par le fabricant. Par ailleurs, lorsqu’une substance classée CMR fait partie d’un dispositif médical, cela doit être notifié sur l’emballage et le patient/consommateur devra en être aussi informé.
Avec la mise en application, le 26 mai 2021, du MDR (UE) 2017/745, la situation des chirurgiens-dentistes va devenir assez inconfortable. L’utilisation fréquente d’alliages dentaires cobalt-chrome (Co-Cr) contenant donc une substance CMR entraînera automatiquement une remise en cause de la pratique professionnelle. Comment en sommes-nous arrivés là ? Quel est l’échéancier auquel nous allons devoir faire face à partir de 2021 ?
Législation européenne sur les produits chimiques
Ces dernières années, l’Union européenne a œuvré pour établir une politique réglementaire commune envers les produits chimiques (fig. 1). Pour cela, il a été publié le règlement (CE) No 1907/2006 du Parlement européen et du Conseil du 18 décembre 2006 concernant l’enregistrement, l’évaluation et l’autorisation des substances chimiques (REACH), ainsi que les restrictions applicables à ces substances, instituant également une agence européenne des produits chimiques (ECHA) [3].
Ce règlement, en vigueur depuis le 1er juin 2007, a pour objectif d’assurer un niveau élevé de protection de la santé humaine et de l’environnement. Différentes échéances importantes ont été fixées et la dernière, celle du 1er juin 2018, a permis sa complète mise en application.
Les entreprises sont responsables du recueil des informations sur les propriétés et de l’utilisation des substances qu’elles fabriquent ou importent en quantités égales ou supérieures à une tonne par an. Elles doivent également évaluer les dangers et les risques que ces substances peuvent présenter. Ces informations sont envoyées à l’ECHA par le biais d’un dossier d’enregistrement. Basé sur le principe « une substance, un enregistrement », l’enregistrement s’applique aux substances et aux substances apparentées telles que celles contenues dans les mélanges et, dans certains cas, celles énumérées dans les articles.
Afin de compléter REACH, un second règlement (CE) No 1272/2008 relatif à la classification, à l’étiquetage et à l’emballage (en anglais, CLP) des substances et des mélanges, a été publié le 16 décembre 2008 [4]. Ce dernier est entré en vigueur le 20 janvier 2009. Il harmonise les critères de classification des substances et des mélanges, ainsi que les règles relatives à l’étiquetage et à l’emballage des substances et mélanges dangereux. Il vise également à établir un inventaire des classifications et des étiquetages des substances enregistrées. Lors de la dernière mise à jour (10 janvier 2021), la base de données contenait 23 121 substances uniques et des informations provenant de 101 732 dossiers. Par exemple, le Cr métallique (EC/231-157-5) et le Co métallique (EC/231-158-0) ont été enregistrés respectivement le 8 et 17 septembre 2010 [5].
Ce règlement CLP a notamment introduit deux catégories de danger (1 et 2) qui définissent le niveau de preuve de l’effet CMR (tab. I) [4]. La catégorie 1 est elle-même divisée en deux sous-catégories (1A-effets avérés et 1B-effets présumés). Seules les substances classées CMR de catégorie 1A/1B sont soumises à restriction par REACH. Cette classification est la seule en vigueur dans l’Union européenne, et donc en France.
Depuis 2009 et la publication de la première « Adaptation au progrès technique » (APT), de nombreuses mises à jour et corrections ont été apportées. Le 18 février 2020, la classification harmonisée du Co métallique a été actée en annexe VI du CLP, et rentrera en vigueur à partir du 9 septembre 2021 [6]. Le Co métallique est une substance C1B, M2 et R1B.
|
Tableau I – Définitions des catégories de dangers « CMR » selon la classification de l’Union européenne |
||
|
Effets |
Catégories |
Définitions des catégories |
|
Cancérogènes |
Catégorie 1A |
Substances dont le potentiel cancérigène pour l’être humain est avéré |
|
Catégorie 1B |
Substances dont le potentiel cancérogène pour l’être humain est supposé |
|
|
Catégorie 2 |
Substances suspectées d’être cancérogènes pour l’homme |
|
|
Mutagènes |
Catégorie 1A |
Substances dont la capacité d’induire des mutations héréditaires dans les cellules germinales des êtres humains est avérée |
|
Catégorie 1B |
Substances dont la capacité d’induire des mutations héréditaires dans les cellules germinales des êtres humains est supposée |
|
|
Catégorie 2 |
Substances préoccupantes du fait qu’elles pourraient induire des mutations héréditaires dans les cellules germinales des êtres humains est supposée |
|
|
toxiques pour la Reproduction |
Catégorie 1A |
Substances dont la toxicité pour la reproduction humaine est avérée |
|
Catégorie 1B |
Substances présumées toxiques pour la reproduction humaine |
|
|
Catégorie 2 |
Substances suspectées d’être toxiques pour la reproduction humaine |
|
Exigences réglementaires relatives aux dispositifs médicaux
Parallèlement, et ce depuis des années aussi, l’UE a pour objectif d’harmoniser la législation des dispositifs médicaux au sein de ses pays membres. En 1993, elle a ainsi publié la directive sur les dispositifs médicaux (DDM 93/42/CEE) [7]. Ensuite, le règlement sur les dispositifs médicaux (RDM) (UE) 2017/745) est entré en vigueur le 26 mai 2017 [2]. Après une période de transition de trois ans, il aurait dû s’appliquer à partir du 26 mai 2020. Néanmoins en raison de la crise sanitaire, la date d’application a été reportée au 26 mai 2021 par une décision publiée le 24 avril 2020, le règlement (UE) 2020/561 modifiant ainsi le règlement (UE) 2017/745 [8].
L’objectif est là aussi une meilleure protection en termes de santé publique et de sécurité des patients. Les dispositifs médicaux sont classés en fonction du niveau de risque associé à leur utilisation (classe I, classe IIA, classe IIB et classe III, selon un risque croissant). Si deux règles ou plus s’appliquent, la classification à conserver est la plus élevée. Le nouveau règlement a revu les règles de classification et en a précisé de nouvelles. Parmi ces nouvelles exigences, il est obligatoire d’indiquer la présence de toute substance classée CMR 1A ou 1B, dans une concentration supérieure à 0,1 % en fraction massique (m/m). Par ailleurs pour les dispositifs à risque élevé, un contrôle plus strict est nécessaire avant leur mise sur le marché. Une nouvelle base de données de l’UE sur les dispositifs médicaux (EUDAMED) est également créée. Un identifiant unique de dispositif (IUD) permettra d’optimiser la traçabilité des dispositifs médicaux. Enfin, dans le cas de dispositifs défectueux, un nouveau mécanisme financier devrait permettre aux patients d’être effectivement indemnisés.
À partir de mars 2021, le premier module de l’EUDAMED pour l’enregistrement des opérateurs économiques (fabricants, représentants autorisés et importateurs) sera disponible. Il permettra à ces opérateurs d’obtenir un numéro d’enregistrement unique (en anglais, SRN) qui donnera accès aux autres fonctionnalités de la base de données. En 2022, la base de données EUDAMED devrait être pleinement opérationnelle.
À compter du 26 mai 2021, tous les dispositifs médicaux ne devront être approuvés qu’en fonction du processus RDM. Les certificats et dispositifs déjà sur le marché resteront valides jusqu’en mai 2022 (certificats de conformité CE délivrés avant le 25 mai 2017) ou jusqu’en mai 2024 (certificats de conformité CE délivrés après le 25 mai 2017).
Nouvelle convention nationale organisant les rapports entre les chirurgiens-dentistes libéraux et l’assurance maladie
La nouvelle convention nationale 2018-2023 organisant les rapports entre les chirurgiens-dentistes libéraux et l’assurance maladie a été publiée le 25 août 2018 [9]. Cette convention prévoit un calendrier d’application et notamment, depuis le 1er janvier 2020, l’entrée du dispositif « paniers », avec un reste à charge 0 (RAC-0) et des tarifs maîtrisés (RAC-M).
Le tableau II rassemble l’ensemble des correspondances entre les termes clés anglais et français, afin de mieux comprendre comment la réglementation européenne s’applique au niveau national.
S’il faut se réjouir que l’article 7-1 de la convention mette en place une différenciation des prothèses fixées suivant le matériau utilisé (céramique monolithique [zircone ou hors zircone], céramo-métallique, métallique, céramo-céramique, résine ou composite), allant même jusqu’à différencier les alliages « précieux » des alliages « non précieux », il peut être regrettable, d’une part, que la désignation de ces derniers ne soit en conformité avec les normes internationales, puisqu’à ce jour les termes précieux et non précieux ne sont plus retenus [10]. De plus, aucune précision n’a été donnée sur la composition attendue, notamment des alliages dits non précieux qui regroupent trois catégories : (i) les alliages nickel-chrome (Ni-Cr) de moins en moins utilisés depuis la publication de l’arrêté du 6 mars 2009 [11] relatif à l’interdiction de la mise sur le marché de certains produits contenant du Ni qui, bien que ne concernant pas le domaine de la santé, a sensibilisé les prothésistes et les chirurgiens-dentistes sur les risques liés à ces alliages ; (ii) les alliages Co-Cr qui dominent actuellement le marché, utilisés depuis fort longtemps en prothèse amovible, sont aussi utilisées en prothèse fixée après modification de leur composition ; et (iii) le titane (Ti), matériau apparu comme prometteur mais dont la mise en œuvre reste très difficile.
Est-il éthique qu’un chirurgien-dentiste puisse prescrire à son patient la mise en place d’une prothèse fabriquée à partir d’un alliage contenant une substance CMR ?
|
Tableau II – Correspondance entre les termes clés anglais et français |
|
|
Termes anglais (abréviation) |
Termes français (abréviation) |
|
European Chemicals Agency (ECHA) |
Agence européenne des produits chimiques |
|
Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) |
Enregistrement, évaluation et autorisation des substances chimiques |
|
Risk Assessment Committee (RAC) |
Comité d’évaluation des risques (CER) |
|
Classification, Labelling and Packaging (CLP) |
Classification, étiquetage et emballage des produits chimiques |
|
Medical Devices Regulation (MDR) |
Règlement des dispositifs médicaux (RDM) |
|
Medical Devices Directive (MDD) |
Directive des dispositifs médicaux (DDM) |
|
Notified Body (NB) |
Organisme notifié (ON) |
|
Unique Device Identification (UDI) |
Identifiant unique de dispositif (IUD) |
|
Single Registration Number (SRN) |
Numéro d’enregistrement unique |
|
Adaptation to Technical Progress (ATP) |
Adaptation au progrès technique (APT) |
|
European database on medical devices (EUDAMED) |
Base de données européenne du dispositif médical |
Conclusion
À partir du 27 mai 2025, le règlement RDM (UE) 2017/745 s’appliquera à tous les dispositifs médicaux [12]. Dans le contexte de l’utilisation d’une substance CMR dans un alliage dentaire et de l’ensemble réglementaire en vigueur (RDM, REACH, CLP), il pourrait être interdit de commercialiser de tels dispositifs médicaux. Ce règlement indique que tout dispositif médical contenant une substance CMR (comme pour les alliages Co-Cr) est conforme à la réglementation si la concentration de la substance CMR 1B (ici, le Co) est inférieure à 0,1 % (m/m). Dans les alliages dentaires Co-Cr, le Co est généralement le composant principal, avec des pourcentages largement supérieurs à 0,1 %. Seule une justification raisonnée qu’aucune substitution n’est possible pourrait amener l’ON en charge du dossier à accorder le marquage CE à un dispositif contenant du Co métallique avec une concentration supérieure à 0,1 % en fraction massique.
A contrario, depuis janvier 2020, la nouvelle convention nationale promeut au sein du panier RAC-0 des éléments prothétiques métalliques et/ou céramo-métalliques qui sont, depuis quelques années, le plus souvent des alliages Co-Cr dont l’élément constitutif majeur est le Co, récemment classé comme substance CMR 1B. Cette politique incitative va à l’encontre des préconisations réglementaires européennes.
Pourrons-nous proposer une alternative satisfaisante avant le mois de mai 2025 ? Ou serons-nous contraints de proposer à nouveau des prothèses en nickel-chrome, le nickel métallique étant seulement classé dans le groupe 2B des substances carcinogènes possibles pour l’Homme [13], échappant, même si c’est de peu, à REACH ?
Christine Janssen
Institut de formation en masso-kinésithérapie pour déficients de la vue (IFMK DV), Lyon
Alina Vaicelyte
LMI CNRS, UMR 5615,
Université Lyon 1
Marc Le Borgne
Small Molecules for Biological Targets Team, Centre de recherche en cancérologie de Lyon,
Centre Léon Bérard, CNRS 5286,
INSERM 1052, Université Lyon 1
Brigitte Grosgogeat
LMI CNRS 5615, Université Lyon 1,
Faculté d’odontologie de Lyon,
Université Lyon 1,
Service d’odontologie,
Hospices Civils de Lyon
brigitte.grosgogeat@univ-lyon1.fr












Commentaires